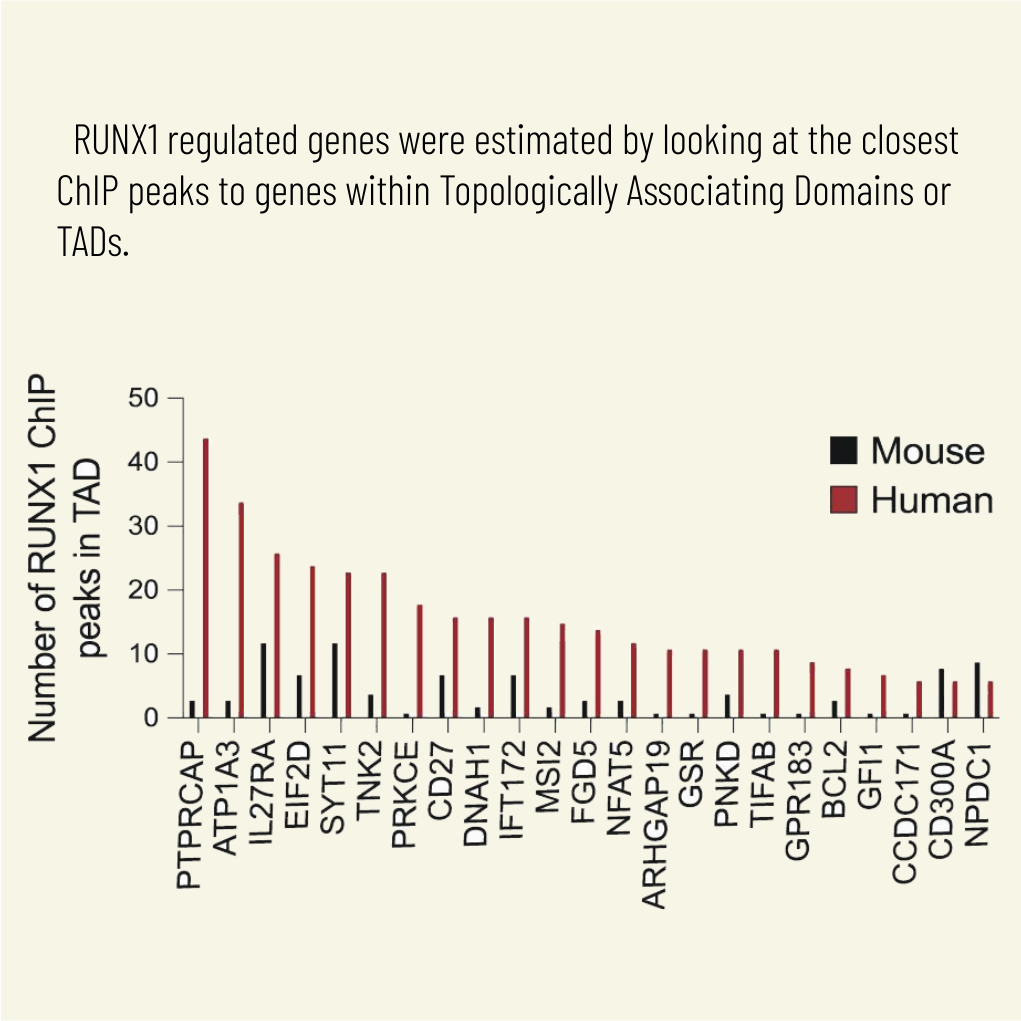
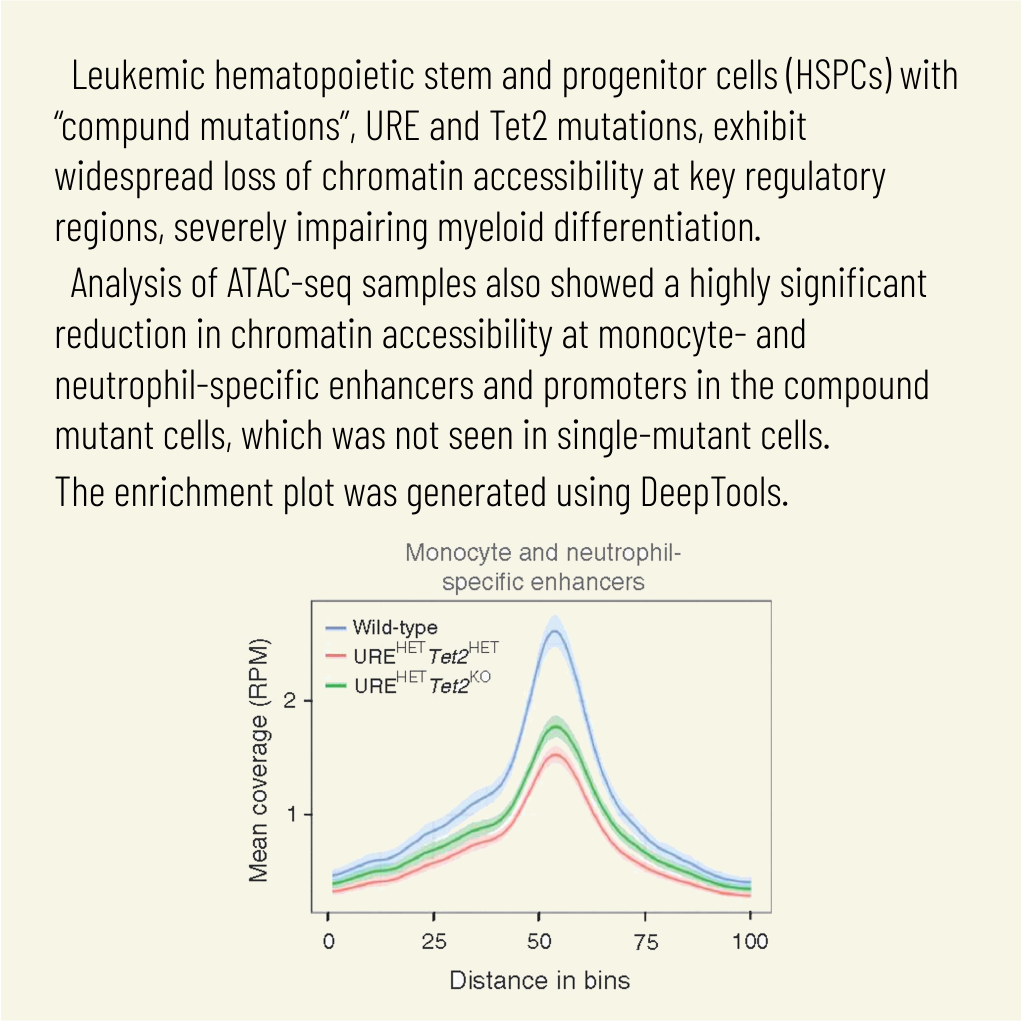
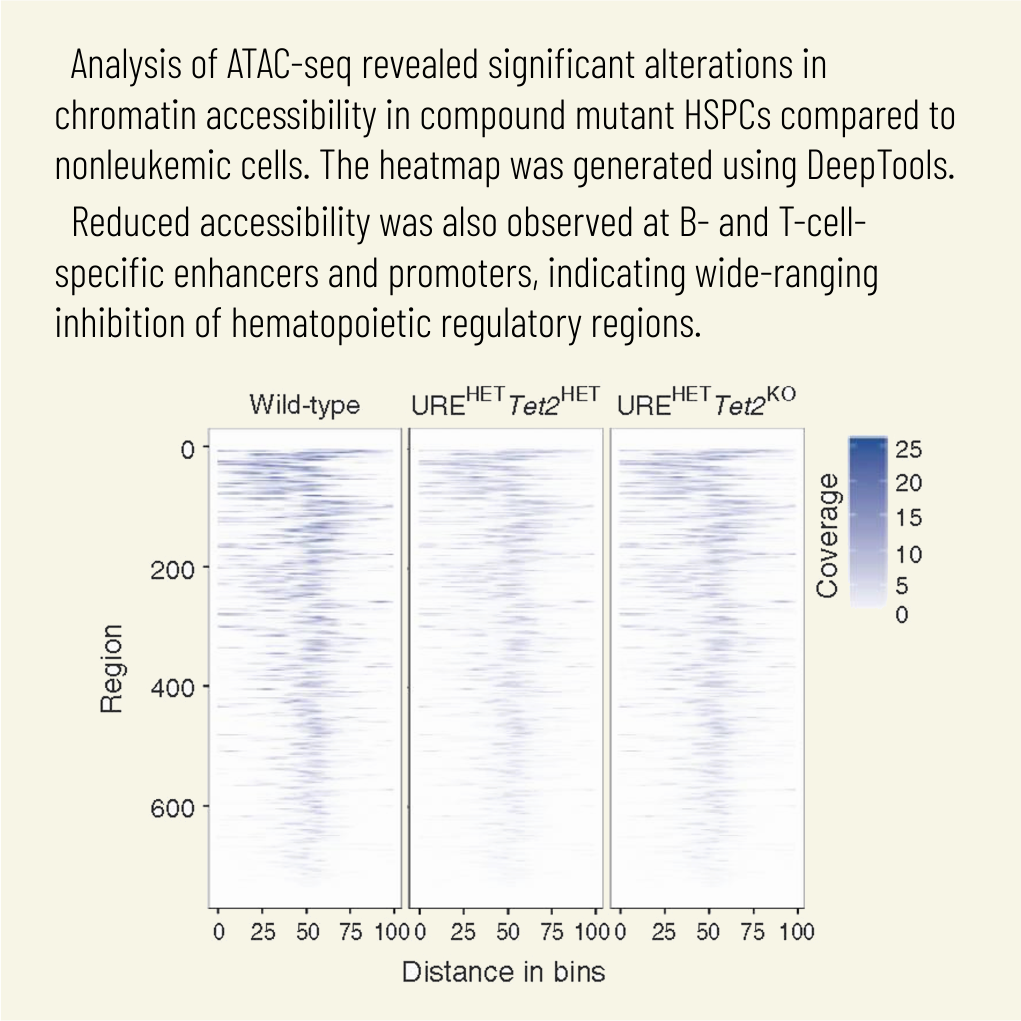
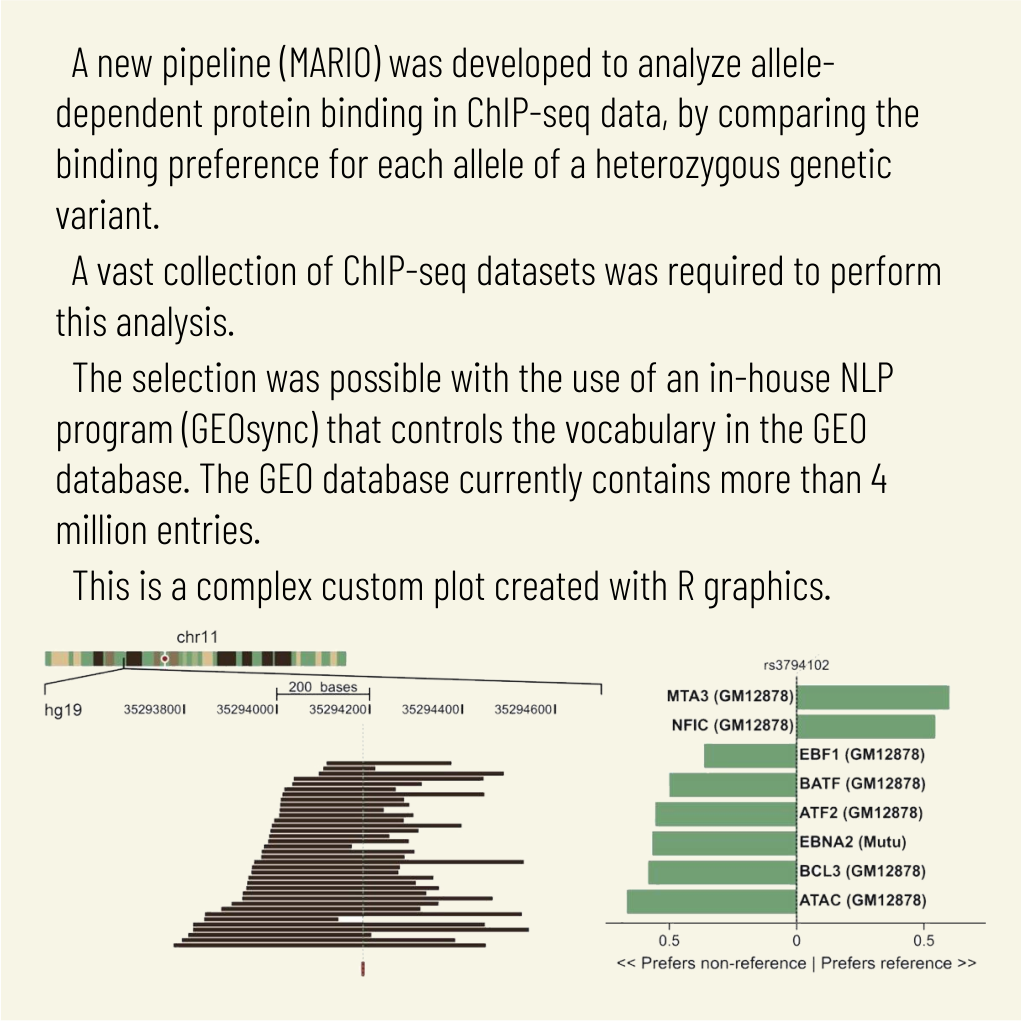
Cell Mol Biol Lett. 2025 Mar 28;30(1):36. doi: 10.1186/s11658-025-00711-z.
Genetic and epigenetic regulation of Treg cell fitness by autism-related chromatin remodeler CHD8.
Jun-Qi Yang 1, Chen Wang 1, Ramesh C Nayak 1, Manohar Kolla 1, Mingjun Cai 1, Mario Pujato 2, Yi Zheng 1, Q Richard Lu 3, Fukun Guo 4
Affiliations:
1 Division of Experimental Hematology and Cancer Biology, Children's Hospital Medical Center, Department of Pediatrics, University of Cincinnati College of Medicine, 3333 Burnet Avenue, Cincinnati, OH, 45229, USA.
2 Life Sciences Computational Services LLC, Huntingdon Valley, PA, 19006, USA.
3 Division of Experimental Hematology and Cancer Biology, Children's Hospital Medical Center, Department of Pediatrics, University of Cincinnati College of Medicine, 3333 Burnet Avenue, Cincinnati, OH, 45229, USA. richard.lu@cchmc.org.
4 Division of Experimental Hematology and Cancer Biology, Children's Hospital Medical Center, Department of Pediatrics, University of Cincinnati College of Medicine, 3333 Burnet Avenue, Cincinnati, OH, 45229, USA. fukun.guo@cchmc.org.
Abstract:
Background: Chromatin remodeler chromodomain helicase DNA-binding protein 8 (CHD8) defines a subtype of autism that is associated with immune disorders. It remains unknown whether CHD8 plays a cell-intrinsic role in immune cells such as regulatory T cells (Tregs) that maintain immune tolerance through suppressing CD4+ and CD8+ effector T cells.
Methods: Treg-specific conditional CHD8-deficient mice were generated by crossing Chd8Flox/Flox mice with Foxp3YFP-cre transgenic mice. Effects of CHD8 deficiency were investigated using hematoxylin and eosin (H&E) staining, flow cytometry, and multi-omics, including RNA-sequencing (RNA-seq), assay for transposase-accessible chromatin sequencing (ATAC-seq), and chromatin immunoprecipitation sequencing (CHIP-seq).
Results: We found that Treg-specific CHD8 deletion led to early, fatal inflammation owing to increased CD4+ and CD8+ effector T cells. CHD8 deletion did not alter Treg homeostasis but increased their functional plasticity with elevated expression of effector T cell cytokines. CHIP-seq of Tregs uncovered that CHD8 binding genes were enriched in phosphatidylinositol-3 kinase (PI3K)-protein kinase B (Akt)-mammalian target of rapamycin (mTOR) signaling and several other pathways. RNA-seq and ATAC-seq revealed that CHD8 deletion upregulated a number of pathways, notably mammalian target of rapamycin complex 1 (mTORC1) signaling and its mediated glycolysis that have been reported to promote Treg plasticity. Integrating RNA-seq data with CHIP-seq and ATAC-seq data identified a number of CHD8 target genes whose expression depends on CHD8 direct binding-mediated chromatin remodeling.
Conclusions: Our findings suggest that CHD8 plays an important role in maintaining Treg fitness through genetic and epigenetic mechanisms to control autoimmunity, which may have important implications in immune changes in autism.
iScience. 2024 Apr 24;27(6):109809. doi: 10.1016/j.isci.2024.109809. eCollection 2024 Jun 21.
Dysregulated innate immune signaling cooperates with RUNX1 mutations to transform an MDS-like disease to AML
Laura Barreyro 1, Avery M Sampson 1, Kathleen Hueneman 1, Kwangmin Choi 1, Susanne Christie 1, Vighnesh Ramesh 1, Michael Wyder 2, Dehua Wang 3 4, Mario Pujato 5, Kenneth D Greis 2, Gang Huang 1 6 7, Daniel T Starczynowski 1 2 8
Affiliations:
1 Division of Experimental Hematology and Cancer Biology, Cincinnati Children's Hospital, Cincinnati, OH, USA.
2 Department of Cancer Biology, University of Cincinnati, Cincinnati, OH, USA.
3 Department of Pathology & Laboratory Medicine, University of Cincinnati, Cincinnati, OH, USA.
4 Department of Pathology, Cincinnati Children's Hospital, Cincinnati, OH, USA.
5 Life Sciences Computational Services, LLC, Huntingdon Valley, PA, USA.
6 Department of Cell Systems & Anatomy, UT Health San Antonio, San Antonio, TX, USA.
7 Department of Pathology & Laboratory Medicine, UT Health San Antonio, San Antonio, TX, USA.
8 University of Cincinnati Cancer Center, Cincinnati, OH, USA.
Abstract:
Dysregulated innate immune signaling is linked to preleukemic conditions and myeloid malignancies. However, it is unknown whether sustained innate immune signaling contributes to malignant transformation. Here we show that cell-intrinsic innate immune signaling driven by miR-146a deletion (miR-146aKO), a commonly deleted gene in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), cooperates with mutant RUNX1 (RUNX1mut) to initially induce marrow failure and features of MDS. However, miR-146aKO hematopoietic stem and/or progenitor cells (HSPCs) expressing RUNX1mut eventually progress to a fatal AML. miR-146aKO HSPCs exhaust during serial transplantation, while expression of RUNX1mut restored their hematopoietic cell function. Thus, HSPCs exhibiting dysregulated innate immune signaling require a second hit to develop AML. Inhibiting the dysregulated innate immune pathways with a TRAF6-UBE2N inhibitor suppressed leukemic miR-146aKO/RUNX1mut HSPCs, highlighting the necessity of TRAF6-dependent cell-intrinsic innate immune signaling in initiating and maintaining AML. These findings underscore the critical role of dysregulated cell-intrinsic innate immune signaling in driving preleukemic cells toward AML progression.
VExD: a curated resource for human gene expression alterations following viral infection
Phillip J Dexheimer 1 2, Mario Pujato 1, Krishna M Roskin 1 3 4, Matthew T Weirauch 4 5 6
Affiliations:
1 Division of Biomedical Informatics, Cincinnati Children's Hospital, Cincinnati, OH 45229, USA.
2 Department of Biomedical Informatics, University of Cincinnati College of Medicine, Cincinnati, OH 45221, USA.
3 Division of Immunobiology, Cincinnati Children's Hospital, Cincinnati, OH 45229, USA.
4 Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH 45221, USA.
5 Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital, Cincinnati, OH 45229, USA.
6 Divisions of Human Genetics, Biomedical Informatics and Developmental Biology, Cincinnati Children's Hospital, Cincinnati, OH 45229, USA.
Abstract:
Much of the host antiviral response is mediated through changes to host gene expression levels. Likewise, viruses induce changes to host gene expression levels in order to promote the viral life cycle and evade the host immune system. However, there is no resource that specifically collects human gene expression levels pre- and post-virus infection. Further, public gene expression repositories do not contain enough specialized metadata to easily find relevant experiments. Here, we present the Virus Expression Database (VExD), a freely available website and database, that collects human gene expression datasets in response to viral infection. VExD contains ∼8,000 uniformly processed samples obtained from 289 studies examining 51 distinct human viruses. We show that the VExD processing pipeline captures known antiviral responses in the form of interferon-stimulated genes. We further show that the datasets collected in VExD can be used to quickly identify supporting data for experiments performed in human cells or model organisms. VExD is freely available at https://vexd.cchmc.org.
Keywords:
RNA-seq; functional genomics; gene expression; human virus; microarray; virus–host interaction; web resource.
Blood Cancer Discov. 2022 Sep 6;3(5):444-467. doi: 10.1158/2643-3230.BCD-21-0226.
PU.1-Dependent Enhancer Inhibition Separates Tet2-Deficient Hematopoiesis from Malignant Transformation
Maria M Aivalioti 1 2, Boris A Bartholdy 1, Kith Pradhan 3, Tushar D Bhagat 3, Aliona Zintiridou 1, Jong Jin Jeong 4, Victor J Thiruthuvanathan 1, Mario Pujato 5, Aditi Paranjpe 5, Chi Zhang 1, Ross L Levine 6, Aaron D Viny 7, Amittha Wickrema 4, Amit Verma 3 8, Britta Will 1 3
Affiliations:
1 Department of Cell Biology, Albert Einstein College of Medicine/Montefiore Medical Center, Bronx, New York.
2 Graduate Programs in the Biomedical Sciences, Albert Einstein College of Medicine, Bronx, New York.
3 Department of Medicine (Oncology), Albert Einstein College of Medicine/Montefiore Medical Center, Bronx, New York.
4 Section of Hematology/Oncology, Department of Medicine, The University of Chicago, Chicago, Illinois.
5 Division of Biomedical Informatics, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio.
6 Center for Hematologic Malignancies, Memorial Sloan Kettering Cancer Center, New York, New York.
7 Department of Genetics and Development, Columbia University, New York, New York.
8 Department of Developmental and Molecular Biology, Albert Einstein College of Medicine/Montefiore Medical Center, Bronx, New York.
Abstract:
Cytosine hypermethylation in and around DNA-binding sites of master transcription factors, including PU.1, occurs in aging hematopoietic stem cells following acquired loss-of-function mutations of DNA methyl-cytosine dioxygenase ten-eleven translocation-2 (TET2), albeit functional relevance has been unclear. We show that Tet2-deficient mouse hematopoietic stem and progenitor cells undergo malignant transformation upon compromised gene regulation through heterozygous deletion of an upstream regulatory region (UREΔ/WT) of the PU.1 gene. Although compatible with multilineage blood formation at young age, Tet2-deficient PU.1 UREΔ/WT mice develop highly penetrant, transplantable acute myeloid leukemia (AML) during aging. Leukemic stem and progenitor cells show hypermethylation at putative PU.1-binding sites, fail to activate myeloid enhancers, and are hallmarked by a signature of genes with impaired expression shared with human AML. Our study demonstrates that Tet2 and PU.1 jointly suppress leukemogenesis and uncovers a methylation-sensitive PU.1-dependent gene network as a unifying molecular vulnerability associated with AML.
Significance: We identify moderately impaired PU.1 mRNA expression as a biological modality predisposing Tet2-deficient hematopoietic stem and progenitor cells to malignant transformation. Our study furthermore uncovers a methylation-sensitive PU.1 gene network as a common feature of myeloid leukemia potentially allowing for the identification of patients at risk for malignant transformation. See related commentary by Schleicher and Pietras, p. 378. This article is highlighted in the In This Issue feature, p. 369.
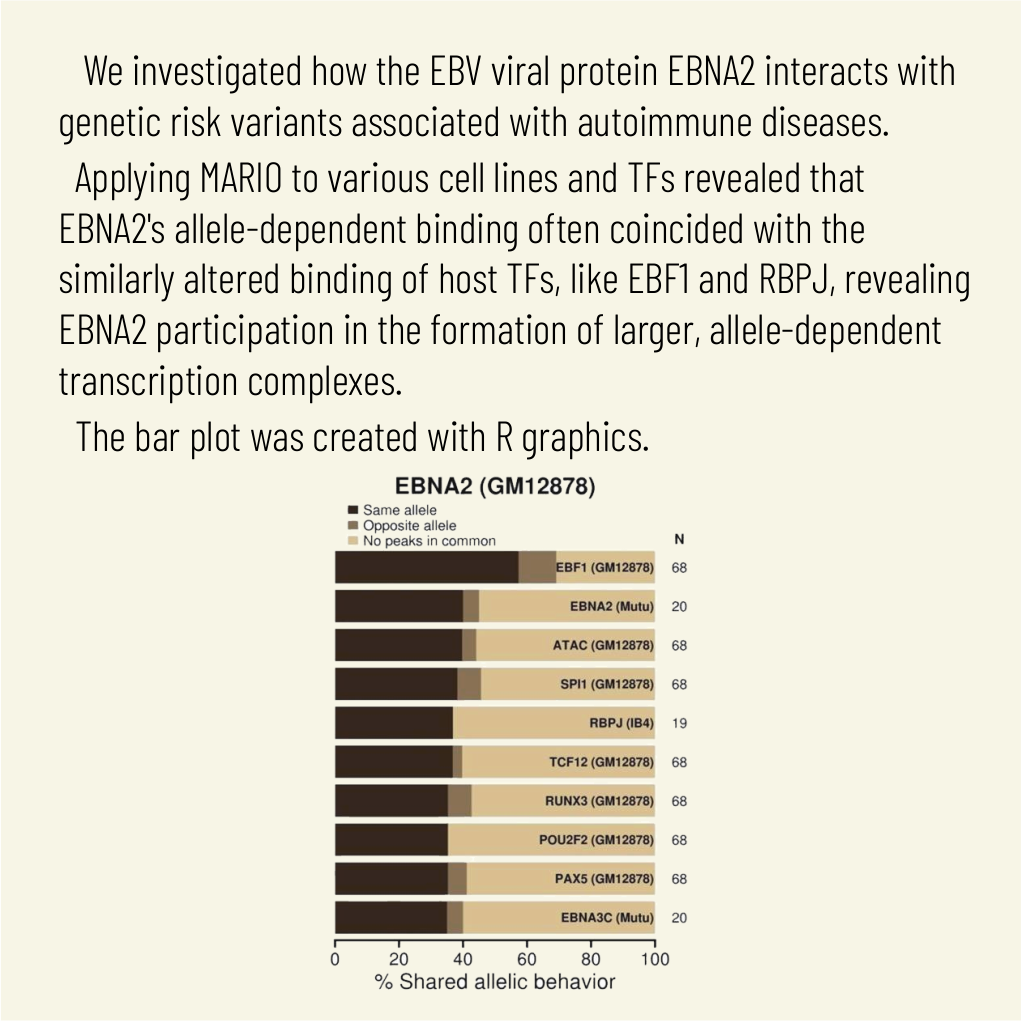
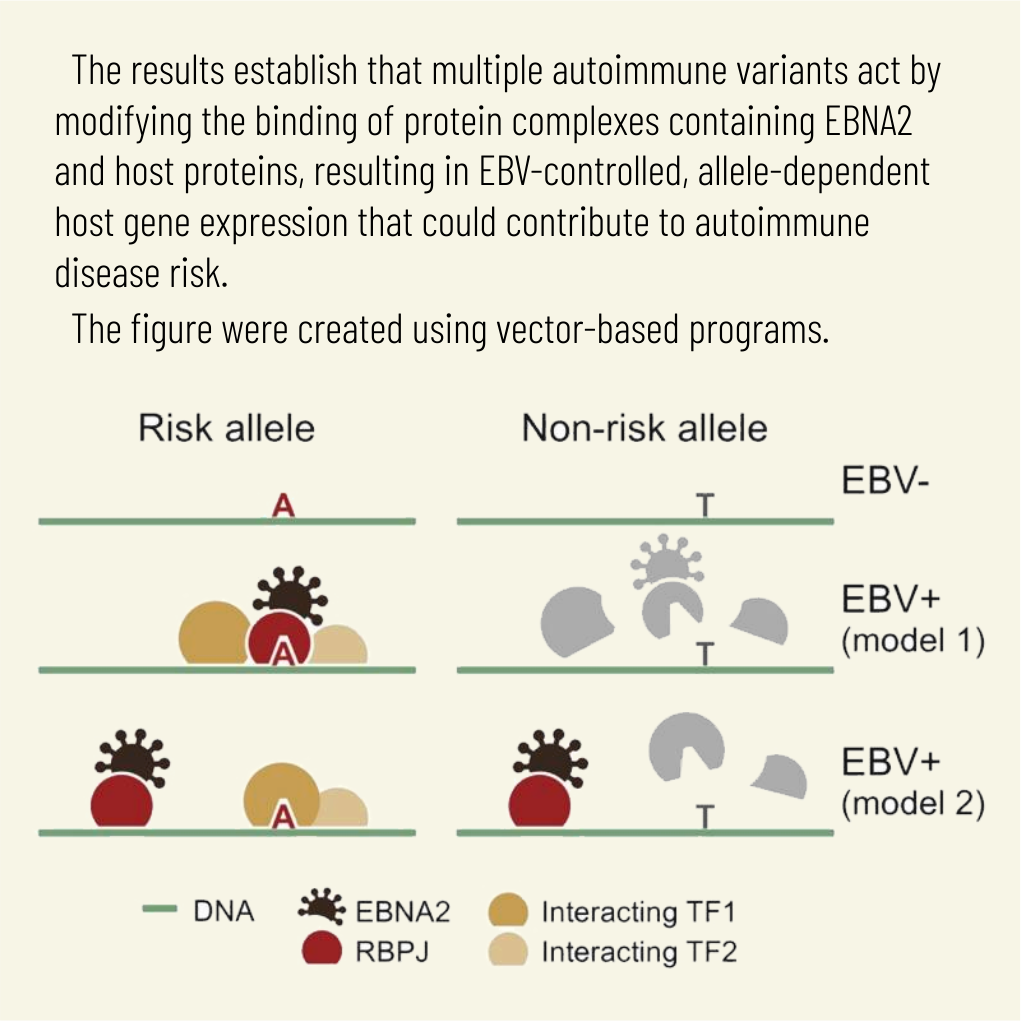
Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity
John B Harley * 1 2 3 4 5, Xiaoting Chen * 6, Mario Pujato * 6, Daniel Miller 6, Avery Maddox 6, Carmy Forney 6, Albert F Magnusen 6, Arthur Lynch 6, Kashish Chetal 7, Masashi Yukawa 8, Artem Barski 9 8 10, Nathan Salomonis 9 7, Kenneth M Kaufman 6 11 9 12, Leah C Kottyan 13 14, Matthew T Weirauch 15 16 17 18
Affiliations:
1 Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. john.harley@cchmc.org.
2 Division of Immunobiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. john.harley@cchmc.org.
3 Division of Developmental Biology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. john.harley@cchmc.org.
4 Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA. john.harley@cchmc.org.
5 US Department of Veterans Affairs Medical Center, Cincinnati, OH, USA. john.harley@cchmc.org.
6 Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
7 Division of Biomedical Informatics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
8 Division of Allergy & Immunology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
9 Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA.
10 Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
11 Division of Immunobiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA.
12 US Department of Veterans Affairs Medical Center, Cincinnati, OH, USA.
13 Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. leah.kottyan@cchmc.org.
14 Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA. leah.kottyan@cchmc.org.
15 Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. matthew.weirauch@cchmc.org.
16 Division of Developmental Biology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. matthew.weirauch@cchmc.org.
17 Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA. matthew.weirauch@cchmc.org.
18 Division of Biomedical Informatics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA. matthew.weirauch@cchmc.org.
* Contributed equally.
Abstract:
Explaining the genetics of many diseases is challenging because most associations localize to incompletely characterized regulatory regions. Using new computational methods, we show that transcription factors (TFs) occupy multiple loci associated with individual complex genetic disorders. Application to 213 phenotypes and 1,544 TF binding datasets identified 2,264 relationships between hundreds of TFs and 94 phenotypes, including androgen receptor in prostate cancer and GATA3 in breast cancer. Strikingly, nearly half of systemic lupus erythematosus risk loci are occupied by the Epstein-Barr virus EBNA2 protein and many coclustering human TFs, showing gene-environment interaction. Similar EBNA2-anchored associations exist in multiple sclerosis, rheumatoid arthritis, inflammatory bowel disease, type 1 diabetes, juvenile idiopathic arthritis and celiac disease. Instances of allele-dependent DNA binding and downstream effects on gene expression at plausibly causal variants support genetic mechanisms dependent on EBNA2. Our results nominate mechanisms that operate across risk loci within disease phenotypes, suggesting new models for disease origins.
Iterative sorting of apical and basolateral cargo in Madin-Darby canine kidney cells
Aleksandr Treyer 1, Mario Pujato 2, Ximo Pechuan 3, Anne Müsch 4
Affiliations:
1 Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY 10461.
2 Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY 10461 Center for Autoimmune Genomics and Etiology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH 45229.
3 Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY 10461.
4 Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY 10461 anne.muesch@einstein.yu.edu.
Abstract:
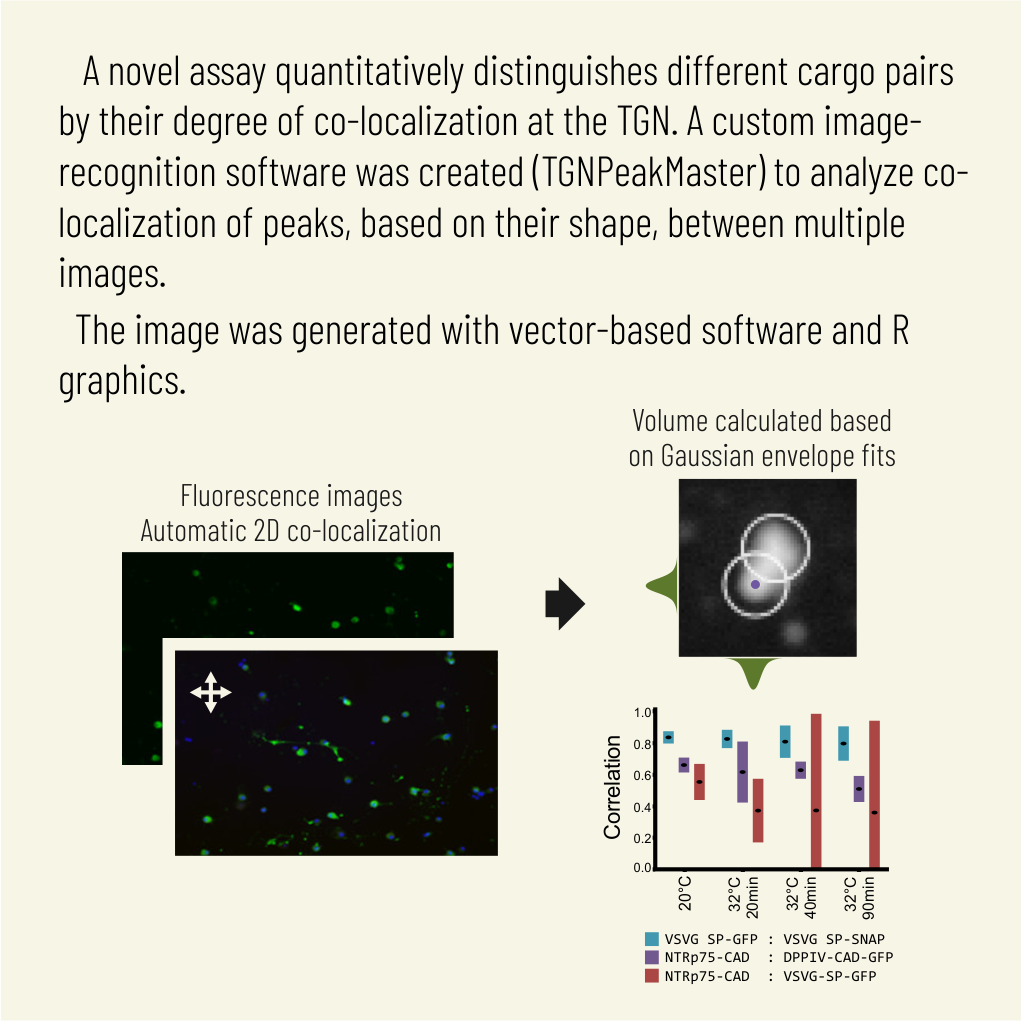
For several decades, the trans-Golgi network (TGN) was considered the most distal stop and hence the ultimate protein-sorting station for distinct apical and basolateral transport carriers that reach their respective surface domains in the direct trafficking pathway. However, recent reports of apical and basolateral cargoes traversing post-Golgi compartments accessible to endocytic ligands before their arrival at the cell surface and the post-TGN breakup of large pleomorphic membrane fragments that exit the Golgi region toward the surface raised the possibility that compartments distal to the TGN mediate or contribute to biosynthetic sorting. Here we describe the development of a novel assay that quantitatively distinguishes different cargo pairs by their degree of colocalization at the TGN and by the evolution of colocalization during their TGN-to-surface transport. Keys to the high resolution of our approach are 1) conversion of perinuclear organelle clustering into a two-dimensional microsomal spread and 2) identification of TGN and post-TGN cargo without the need for a TGN marker that universally cosegregates with all cargo. Using our assay, we provide the first evidence that apical NTRp75 and basolateral VSVG in Madin-Darby canine kidney cells still undergo progressive sorting after they exit the TGN toward the cell surface.
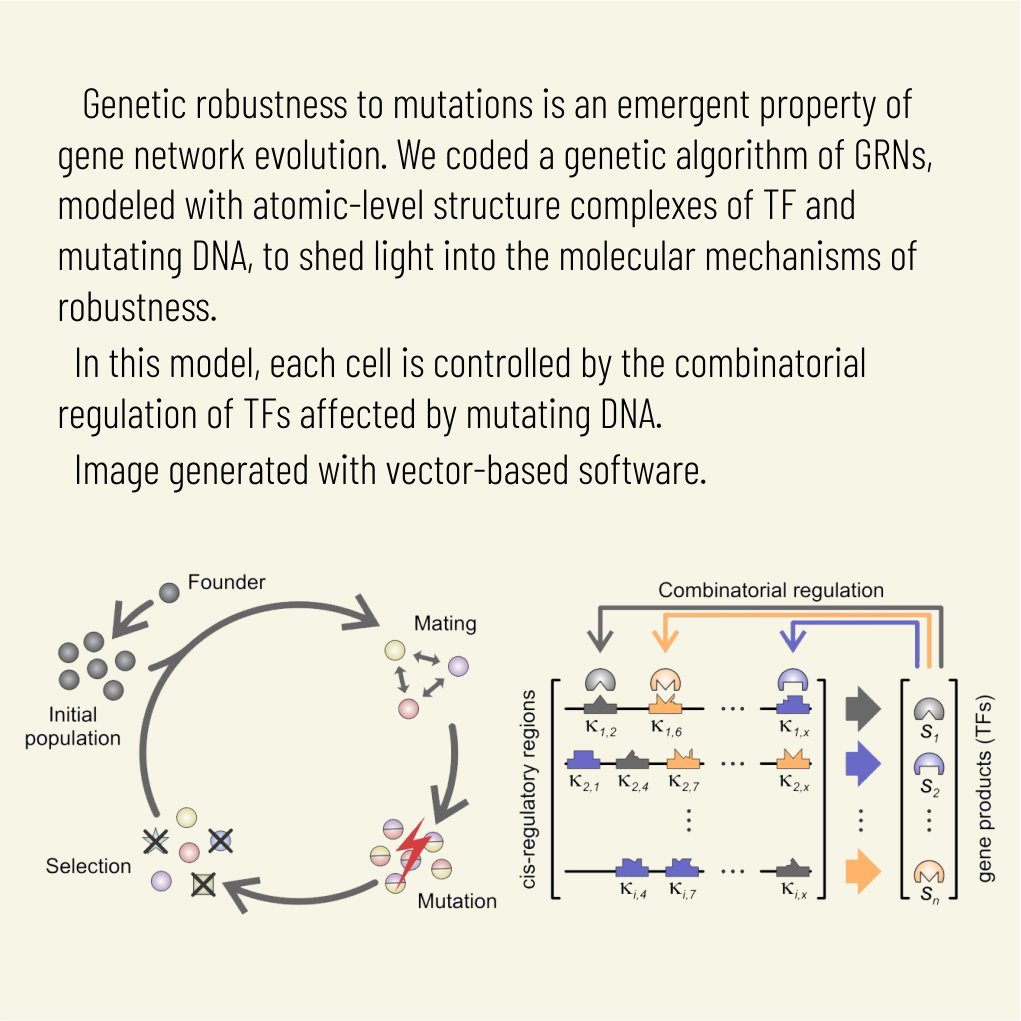
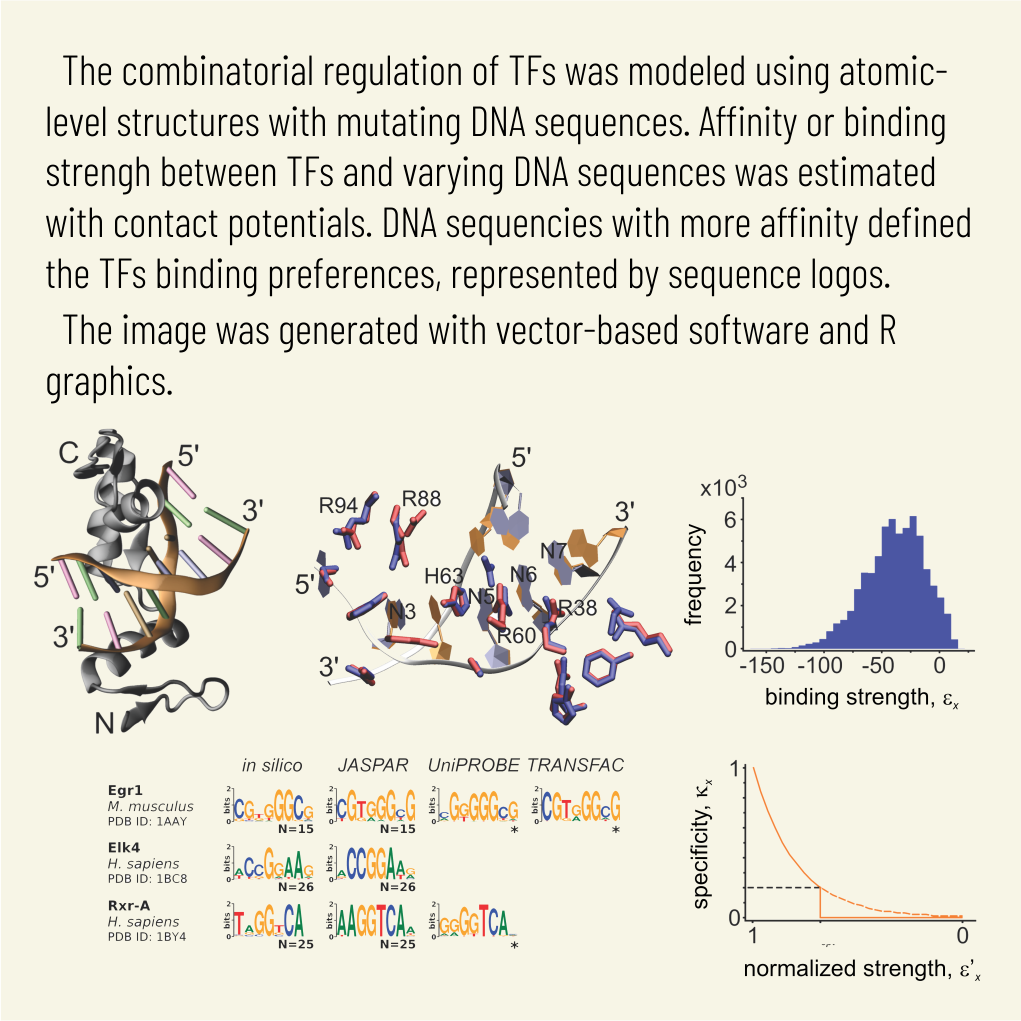
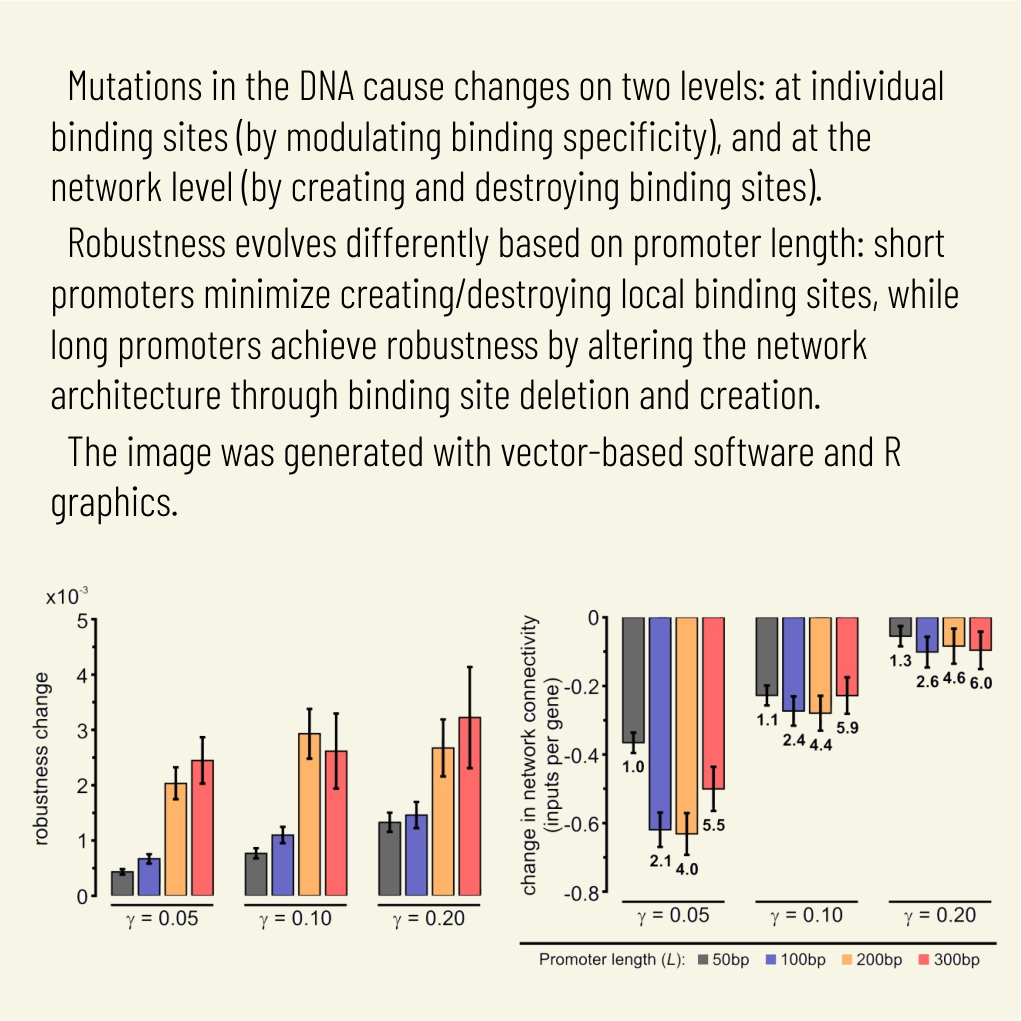
Abstract:
Gene regulatory networks show robustness to perturbations. Previous works identified robustness as an emergent property of gene network evolution but the underlying molecular mechanisms are poorly understood. We used a multi-tier modeling approach that integrates molecular sequence and structure information with network architecture and population dynamics. Structural models of transcription factor-DNA complexes are used to estimate relative binding specificities. In this model, mutations in the DNA cause changes on two levels: (a) at the sequence level in individual binding sites (modulating binding specificity), and (b) at the network level (creating and destroying binding sites). We used this model to dissect the underlying mechanisms responsible for the evolution of robustness in gene regulatory networks. Results suggest that in sparse architectures (represented by short promoters), a mixture of local-sequence and network-architecture level changes are exploited. At the local-sequence level, robustness evolves by decreasing the probabilities of both the destruction of existent and generation of new binding sites. Meanwhile, in highly interconnected architectures (represented by long promoters), robustness evolves almost entirely via network level changes, deleting and creating binding sites that modify the network architecture.